М’язова дистрофія Дюшенна
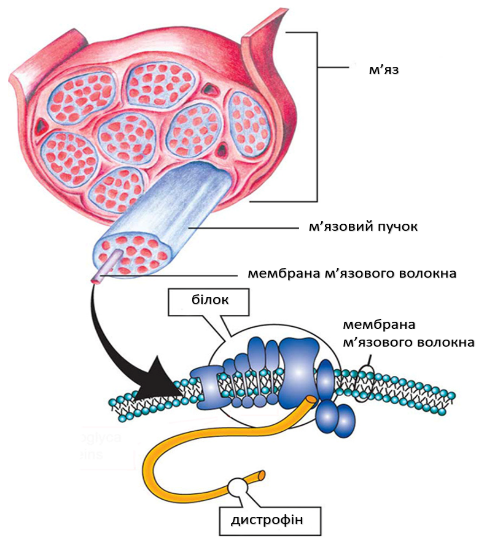
М’язова дистрофія Дюшенна (МДД) – це генетичне захворювання, що характеризується прогресуючою дегенерацією м’язів та їх слабкістю, в результаті генетичного дефекту білка дистрофіну, який допомагає зберігати м’язові клітини неушкодженими.
МДД – одне з чотирьох станів, відомих як дистрофінопатії. Симптоми МДД проявляються в ранньому дитинстві, зазвичай у віці від 2 до 3 років, до цього віку руховий розвиток дитини нормальний. Захворювання в першу чергу вражає хлопчиків, але в рідкісних випадках може вражати і дівчаток.
Поширеність МДД становить приблизно 6 на 100 000 чоловік.
Спадкування при МДД
МДД успадковується за Х-зчепленим типом, тому що ген, який може нести DMD мутацію, знаходиться на Х-хромосомі. Кожен хлопчик успадковує Х-хромосому від матері і Y-хромосому від батька. Дівчата отримують дві Х-хромосоми, по одній від кожного з батьків.
Кожен син, народжений від жінки з мутацією дистрофіна в одній з її двох Х-хромосом, має 50-відсотковий шанс успадкувати дефектний ген і захворіти МДД. Кожна з її дочок має 50-відсотковий шанс успадкувати мутацію і стати носієм. Носії можуть не мати ніяких симптомів хвороби, але можуть мати дітей з мутацією або захворюванням.
Хоча МДД часто зустрічається в сім’ї, в анамнезі якої не було МДД, тобто раптово може народитися син з цим захворюванням. Існує два пояснення цьому. По-перше, генетична мутація, яка призводить до МДД, могла існувати у жінок в сім’ї протягом кількох поколінь, про що ніхто не знав. Можливо, жодна дитина чоловічої статі не народилася з цією хворобою, або, навіть якщо хлопчик в більш ранньому поколінні був вражений, родичі могли не знати, яке у нього захворювання.
Друга можливість полягає в тому, що у дитини з МДД є нова генетична мутація, що виникла в одній з яйцеклітин його матері. Оскільки цієї мутації немає в клітинах крові матері, її неможливо виявити стандартним тестуванням на носійство.
Чоловік з МДД не може передати дефектний ген своїм синам, бо він дає синові Y-хромосому, а не X. Але він обов’язково передасть її своїм дочкам, тому що кожна дочка успадковує єдину X-хромосому свого батька. Тоді вони стануть носіями, і у кожного з їхніх синів буде 50-відсоткова ймовірність розвитку хвороби.
Причина МДД
МДД була вперше описана французьким неврологом Гійомом Бенджаміном Амандом Дюшенна в 1860-х роках. У 1986 році було виявлено ген в Х-хромосомі, який в разі дефекту (мутації) призводить до МДД. У 1987 році був ідентифікований білок, пов’язаний з цим геном, і названий дистрофіном. Ген дистрофіна є найбільшим геном, ідентифікованим у людей, і розташований в короткому плечі Х-хромосоми в локусі Xp21.2.
М’язова дистрофія Дюшенна виникає через те, що мутований ген DMD не може продукувати практично будь-який функціональний дистрофін. Люди з генетичними мутаціями м’язовою дистрофією Беккера виробляють частково функціональний дистрофін. Недолік білка дистрофина в м’язових клітинах робить їх крихкими і легко пошкоджуються.
МДД має рецесивний характер успадкування, зчеплений з Х-хромосомою, і передається від матері, яку називають носієм. Носії МДД – жінки з нормальним геном дистрофина на одній Х-хромосомі і аномальним геном дистрофина на інший Х-хромосомі. Більшість носіїв МДД не мають ознак і симптомів хвороби, але деякі мають ознаки хвороби. Симптоми можуть варіюватися від легкої слабкості скелетних м’язів або ураження серця до важкої слабкості або серцевих порушень і можуть початися в дитинстві або в дорослому віці.
До відносно недавнього часу хлопчики з МДД зазвичай не доживали до підліткового віку. Завдяки досягненням в області кардіологічної та респіраторної допомоги, тривалість життя збільшується, і багато молодих людей з МДД відвідують коледж, роблять кар’єру, одружуються і заводять дітей. Дожити до 30-х років стає все більше, ніж раніше.
 Симптоми МДД
Симптоми МДД
М’язова слабкість – головний симптом МДД. Захворювання може початися вже у віці 2 або 3 років, спочатку вражаючи проксимальні м’язи, а потім вражаються дистальні м’язи кінцівок. У дитини можуть бути труднощі зі стрибками, бігом і ходьбою. Інші симптоми включають збільшення литкових м’язів, хода перевальцем (качина хода) і посилення поперекового лордозу (викривлення хребта всередину). Надалі уражаються також серцевий та дихальні м’язи. Прогресуюча слабкість і сколіоз призводять до порушення функції легенів, що в кінцевому підсумку може викликати гостру дихальну недостатність.
У хлопчиків з МДД часто збільшуються литкові м’язи – псевдогіпертрофія, можуть збільшуватися так само і м’язи стегон.
Слабкість, пов’язана з м’язовою дистрофією Дюшенна, вибірково вражає м’язи кінцівок, розташовані близько до тулуба (проксимальні м’язи), також ноги вражаються раніше рук. Швидкість росту дитини при МДД в перші роки життя зазвичай повільніше, що призводить до низькорослості.
Хлопчики з МДД часто пізно починають ходити. До шкільного віку може здаватися, що дитина незграбна, часто падає. Батьки також можуть помітити, що дітям складно підніматися по сходах, вставати з підлоги або бігати. Піднімаючись з підлоги, хлопчики можуть використовувати опору для рук, щоб піднятися в вертикальне положення.
До шкільного віку діти можуть ходити навшпиньки або подушечках ніг, злегка кульгаючи ходою, і часто падають. Щоб зберегти рівновагу, вони можуть випинати живіт і розправляти плечі. Діти також зазнають труднощів з підняттям рук.
Багато дітей з МДД починають користуватися інвалідним візком приблизно до 12 років. Перехід на інвалідний візок зазвичай відбувається поступово; на перших порах стілець може знадобитися тільки для подолання великих відстаней. Діти часто знову знаходять самостійність, коли повністю переходять на інвалідний візок з електроприводом.
Пацієнти з МДД часто помирають в підлітковому віці або у віці 20 років від дихальної недостатності або кардіоміопатії; лише деякі пацієнти з МДД виживають після третього десятиліття.
Жінки і МДД
Хвороби, успадковані по X-зчепленим рецесивним типом, в основному вражають чоловіків, тому що друга Х-хромосома зазвичай захищає жінок від проявів.
Коли дівчинка успадковує дефектний ген дистрофіна від одного з батьків, вона зазвичай також отримує здоровий ген дистрофіна від другого з батьків, даючи їй досить білка, щоб захистити її від хвороби. Чоловіки, що успадкували мутацію, хворіють, тому що у них немає другого гена дистрофіна, який міг би заповнити дефектний.
У цих жінок дефіцит дистрофина може привести до ослаблення м’язів спини, ніг і рук, які швидко втомлюються. У явних носіїв можуть бути проблеми із серцем, які можуть проявлятися у вигляді задишки або нездатності виконувати помірні вправи. Проблеми з серцем, якщо їх не лікувати, можуть бути досить серйозними і навіть небезпечними для життя.
У дуже рідкісних випадках у дівчинки може повністю бути відсутнім друга Х-хромосома, або її друга Х-хромосома могла бути серйозно пошкоджена. У цих випадках у неї виробляється мало або зовсім відсутній дистрофін (в залежності від типу мутації дистрофина), і у неї розвивається дистрофінопатія, як у хлопчика.
Родичка хлопчика з МДД може пройти повний спектр діагностичних тестів, щоб визначити її статус носійства. Якщо буде встановлено, що вона є носієм МДД, регулярні обстеження сили і ретельний моніторинг серця можуть допомогти їй справитися з будь-якими симптомами, які можуть виникнути.
Біль і чутливість
Зменшення м’язової маси при МДД саме по собі звичайно не викликає хворобливих відчуттів. Деякі люди іноді повідомляють про м’язових судомах.
Оскільки м’язова дистрофія не впливає на нерви безпосередньо, чутлива сфера не страждає. Також не страждає контроль над гладкими або мимовільними м’язами сечового міхура і кишечника, а також сексуальні функції.
Серце
Недолік дистрофина може послабити серцевий м’яз (міокард), що призведе до стану, званому кардіоміопатією. МДД також може викликати порушення провідності в серці. Згодом, іноді вже в підлітковому віці, пошкодження серця, нанесене МДД, може стати небезпечним для життя. Слід уважно стежити за серцем, зазвичай у дитячого кардіолога.
Респіраторна функція
Послідовний контроль дихальної функції слід починати у віці 5-6 років. Діафрагма і інші м’язи, що керують легкими, можуть ослабнути, що зробить легкі менш ефективними. Хоча дитина може не скаржитися на задишку, проблеми, що вказують на погану дихальну функцію, включають головні болі, труднощі з концентрацією уваги або неспання, а також кошмари. Діти, прикуті до інвалідного візка, зазвичай мають ознаки поганої легеневої функції.
Ослаблені дихальні м’язи ускладнюють кашель, що збільшує ризик серйозної респіраторної інфекції. Проста застуда може швидко перейти в пневмонію.
Здатність до навчання
Близько третини хлопчиків з МДД мають деяку ступінь порушення здатності до навчання, хоча деякі мають серйозні когнітивні порушення. Лікарі вважають, що дістрофінове порушення в головному мозку можуть незначно впливати на пізнання і поведінку. Проблеми з навчанням при МДД виникають в трьох основних сферах: концентрація уваги, вербальне навчання і пам’ять і емоційна взаємодія.
Діагностика
При діагностиці будь-якої форми м’язової дистрофії лікар зазвичай починає з вивчення історії хвороби пацієнта і його сім’ї і проведення фізичного обстеження. Лікарі можуть виявити псевдогіпертрофії, посилення поперекового лордозу, порушення ходи і зниження м’язових рефлексів.
Анамнез пацієнта і його фізичний стан мають велико значення для постановки діагнозу, навіть до того, як будуть виконані будь-які складні діагностичні тести.
Кардіоміопатія у пацієнтів з МДД також може бути пов’язана з порушеннями провідності. Лікар може помітити характерні зміни на електрокардіограмі. Крім того, за допомогою ехокардіографії можна виявити структурні зміни в серці, такі як порок клапанів серця (особливо вражає митральний клапан, коли він виникає). Тому необхідні електрокардіограма, неінвазивна візуалізація з ехокардіографії або МРТ серця, а також консультація кардіолога.
Рівні КK і інших ферментів
На початку діагностичного процесу лікарі часто призначають аналіз крові, званий КК (КФК) – креатин (фосфо) киназа, фермент, який виділяється з пошкоджених м’язів. Коли в зразку крові виявляється підвищений рівень КФК, це зазвичай означає, що м’яз руйнується в результаті якого-небудь аномального процесу, такого як м’язова дистрофія або запалення. Дуже високий рівень КK передбачає, що самі м’язи (а не нерви, які їх контролюють) є ймовірною причиною слабкості, хоча це не вказує на те, який саме тип м’язового розлади може мати місце. Високий рівень КФК може бути виявлений до появи симптомів навіть у новонароджених, які страждають МДД.
Рівень КК досягає піку (в 10-20 разів вище верхнього граничного значення) до 2 років, потім поступово падає і в кінцевому підсумку повертається до нормального рівня, коли значну кількість м’язової тканини замінюється жиром і фіброзної тканиною.
Генетичне тестування
Генетичне тестування включає аналіз ДНК будь-яких клітин (зазвичай використовуються клітини крові), щоб побачити, чи є мутація в гені дистрофина, і якщо так, то де саме вона виникає.
Зазвичай генетична діагностика показана пацієнтам з підвищеними рівнями КФК в сироватці і клінічними проявами дистрофінопатії. Діагноз підтверджується, якщо виявлена мутація гена DMD. Генетичний аналіз в першу чергу спрямований на виявлення великих делеційних / дуплікаційних мутацій (в 70-80% випадків присутні такі мутації). Якщо первинний генетичний аналіз негативний, наступним йде аналіз невеликих генних мутацій і генних мікроделецій / дуплікацій.
Родички чоловіків і хлопчиків з МДД можуть пройти ДНК-тестування, щоб визначити, чи є вони носіями хвороби. Жінки, які є носіями МДД, можуть передати хворобу своїм синам, а їх статус носія – своїм дочкам. У меншості випадків дівчинки і жінки, які є носіями МДД, можуть самі виявляти симптоми МДД, такі як м’язова слабкість і проблеми з серцем. Ці симптоми можуть не з’явитися до дорослого віку.
Кілька експериментальних препаратів, які в даний час розробляються для лікування МДД, вимагають знання точної генетичної мутації людини, тому генетичне тестування стало важливим не тільки для діагностики, але, можливо, і для майбутнього лікування.
Біопсія м’язів
Для отримання більш детальної інформації, лікар може призначити м’язову біопсію, хірургічне видалення невеликої вибірки м’язів від пацієнта. Вивчаючи цей зразок, лікарі можуть багато розповісти про те, що насправді відбувається всередині м’язів. Однак в сучасну епоху біопсія м’язів потрібно рідко, тому що майже всім пацієнтам ставиться діагноз генетичного тестування.
Терапія
Ліки, що зменшують навантаження на серце, іноді призначають при МДД.
Ліки, що належать до групи, відомої як кортикостероїди, є основою фармакологічного лікування, оскільки вони виявилися ефективними в уповільненні течії МДД. Дітям слід починати прийом цих ліків до того, як почнеться їх фізичне навантаження.
Кортикостероїди преднізолон і дефлазакорт корисні при лікуванні МДД. FDA 9 лютого 2017 р схвалив дефлазакорт (торгова марка Emflaza), похідне оксазоліна преднізолона, для лікування МДД. Активність 1 мг преднізолону приблизно еквівалентна 1,3 мг дефлазакорта.
Кілька досліджень всіх цих препаратів при МДД показали значне збільшення сили (11% при прийомі преднізолону в порівнянні з плацебо). Це збільшення сили досягло максимуму після трьох місяців лікування і зберігалося протягом 18 місяців. Крім того, були докази поліпшення функції м’язів (наприклад, час, необхідний для підйому на 4 сходинки, на 43% швидше з преднізолоном у порівнянні з плацебо) і легеневої функції. Кортикостероїди також знижують ризик сколіозу і затримують втрату здатності пересуватися. Три дослідження показали, що лікування глюкокортикостероїдами було пов’язано з поліпшенням виживаності. Однак четверте дослідження не показало чіткого зв’язку зі збільшенням виживання.
Також було показано, що легенева функція поліпшується при лікуванні преднізолоном в порівнянні з плацебо. Форсована життєва ємкість легень (FVC) значно покращилася (11%) після шести місяців щоденного лікування преднізолоном.
Глюкокортикоїди можуть затримувати розвиток сколіозу і зменшувати необхідність хірургічного втручання для корекції сколіозу у пацієнтів з МДД. Ризик розвитку сколіозу може бути значно нижче у пацієнтів, які отримують щоденне лікування дефлазакортом, в порівнянні з плацебо, а необхідність в хірургії хребта також значно знижується у пацієнтів, що приймають дефлазакорт. Поширеність переломів у пацієнтів, які отримують глюкокортикоїди, і тих, хто їх не лікує, однакова. Є деякі свідчення того, що лікування глюкокортикоїдами при МДД покращує виживаність, проте інші дані не показують зв’язку між виживанням і лікуванням глюкокортикоїдами.
Постійне застосування кортикостероїдів є частиною стандартної медичної допомоги при МДД, але таке лікування може призвести до побічних ефектів, таких як збільшення ваги, низький зріст, вугри, зміни поведінки, остеопороз, компресійні переломи довгих кісток і хребців. Доречно контролювати пацієнтів, які отримують кортикостероїдну терапію, з періодичною візуалізацією хребта, оскільки вони можуть протікати безсимптомно. Дітей, у яких розвиваються переломи хребців або довгих кісток, слід направляти до дитячого ендокринолога або фахівця по кістках.
Швидке скасування кортикостероїдів може призвести до небезпечних для життя ускладнень.
У вересні 2016 року Управління з санітарного нагляду за якістю харчових продуктів і медикаментів США (FDA) надало прискорене схвалення препарату Етеплірсен, препарату для пропуску екзона, який, як було показано, підвищує рівень дистрофина у пацієнтів з мутацією гена дистрофина, піддається пропуску екзона 51.
Аталурен (також відомий як PTC124) – це пероральний препарат, що розробляється для лікування генетичних дефектів, викликаних безглуздими мутаціями, що дозволяє обійти безглузду мутацію і продовжити процес трансляції до виробництва функціонуючого білка, що було продемонстровано в декількох дослідженнях. Такий підхід може принести користь приблизно від 10% до 15% пацієнтів з МДД / МДБ, які мають безглузді (стоп-мутації) мутації. Аталурен ліцензований в Європейському Союзі і Великобританії для лікування пацієнтів у віці 2 років і старше з МДД, викликаним безглуздими мутаціями.
Найбільш частим побічним ефектом аталурена є блювота. Інші включають зниження апетиту, втрату ваги, головний біль, гіпертонію, кашель, кровотечу з носа, нудоту, біль у верхній частині живота, метеоризм, дискомфорт у животі, запор, висип, біль в кінцівках, скелетно-м’язовий біль в грудях, кров в сечі, мимовільне сечовипускання і лихоманку.
У грудні 2019 року Vyondys 53, препарат для «пропуску екзона», був схвалений FDA для лікування осіб з підтвердженою мутацією гена DMD, піддається терапевтичної стратегії під назвою екзон 53 і може допомогти до 8% людей з МДД.
У серпні 2020 року Viltepso, препарат для «пропуску екзону», був схвалений FDA для лікування людей з підтвердженою мутацією гена DMD, піддається терапевтичній стратегії під назвою екзон 53.